嗜铬细胞瘤
pheochromocytoma
同义词(或曾用名): 副神经节瘤
概述:
起源于肾上腺髓质嗜铬细胞的神经嵴起源的肿瘤
发病部位: 肾上腺
诊断要点:
1, 大多数嗜铬细胞瘤过度生产和释放儿茶酚胺的从而导致相应的临床症状,超过 95%的功能性嗜铬细胞瘤患者呈现不同的迹象持续性或阵发性高血压,而其余均为血压正常。
2,经典的三联征临床表现为头痛,心动过速,大约 25%的患者可出现出汗,不到一半的患者只有这些症状中的一种。其他非特异性症状包括体位性低血压,苍白,震颤,便秘和心理症状,包括急性焦虑和惊慌等;
3,功能性嗜铬细胞瘤可能也会引起内分泌副肿瘤综合征(PNSs)。 最常见的的 PNS 是库欣综合症的驱动因素促肾上腺皮质激素或促肾上腺皮质激素释放激素分泌。 水样腹泻,低血钾,和胃酸缺乏综合症引起的血管活性肠肽的产生和红细胞增多症,由促红细胞生成素(EPO)过度分泌引起或对 EPO 受体过敏也可发生;
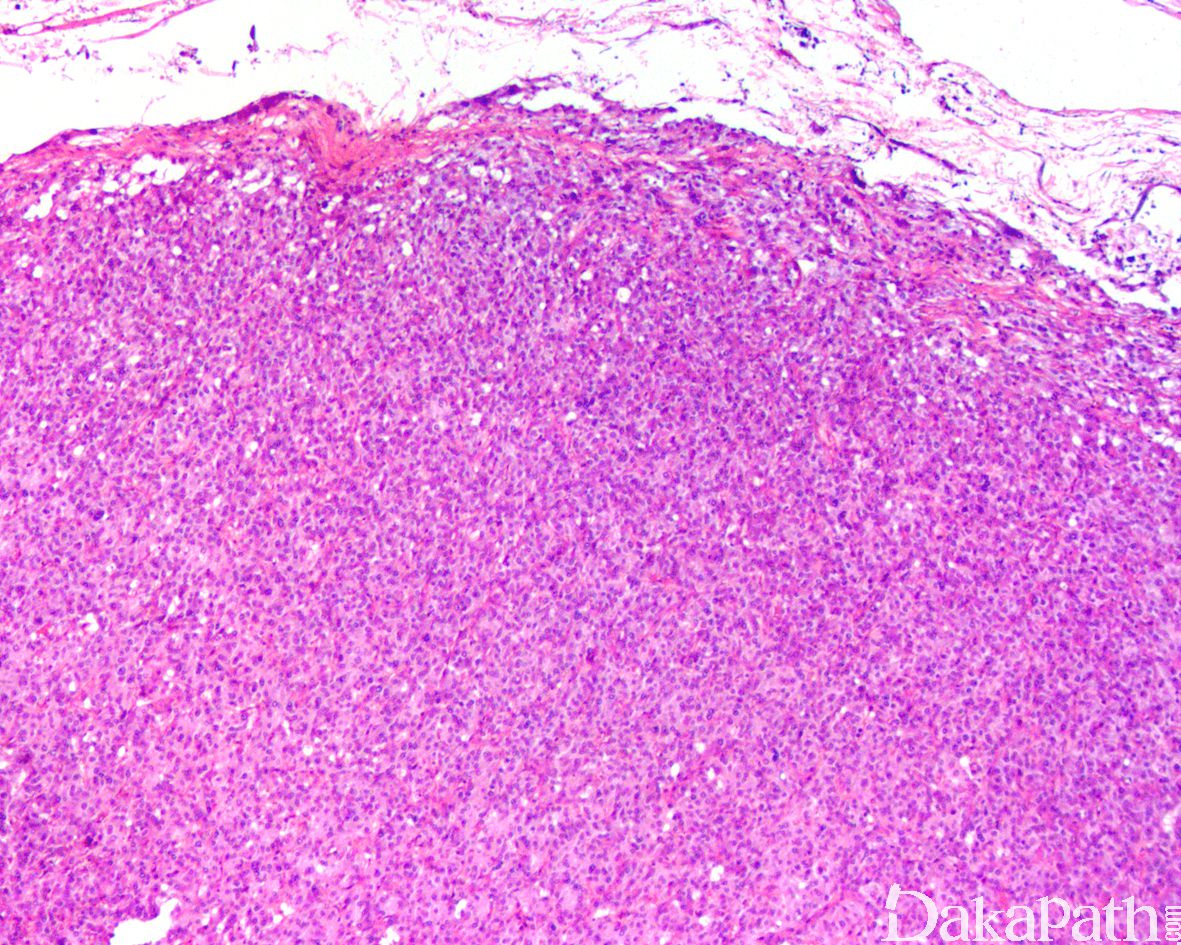
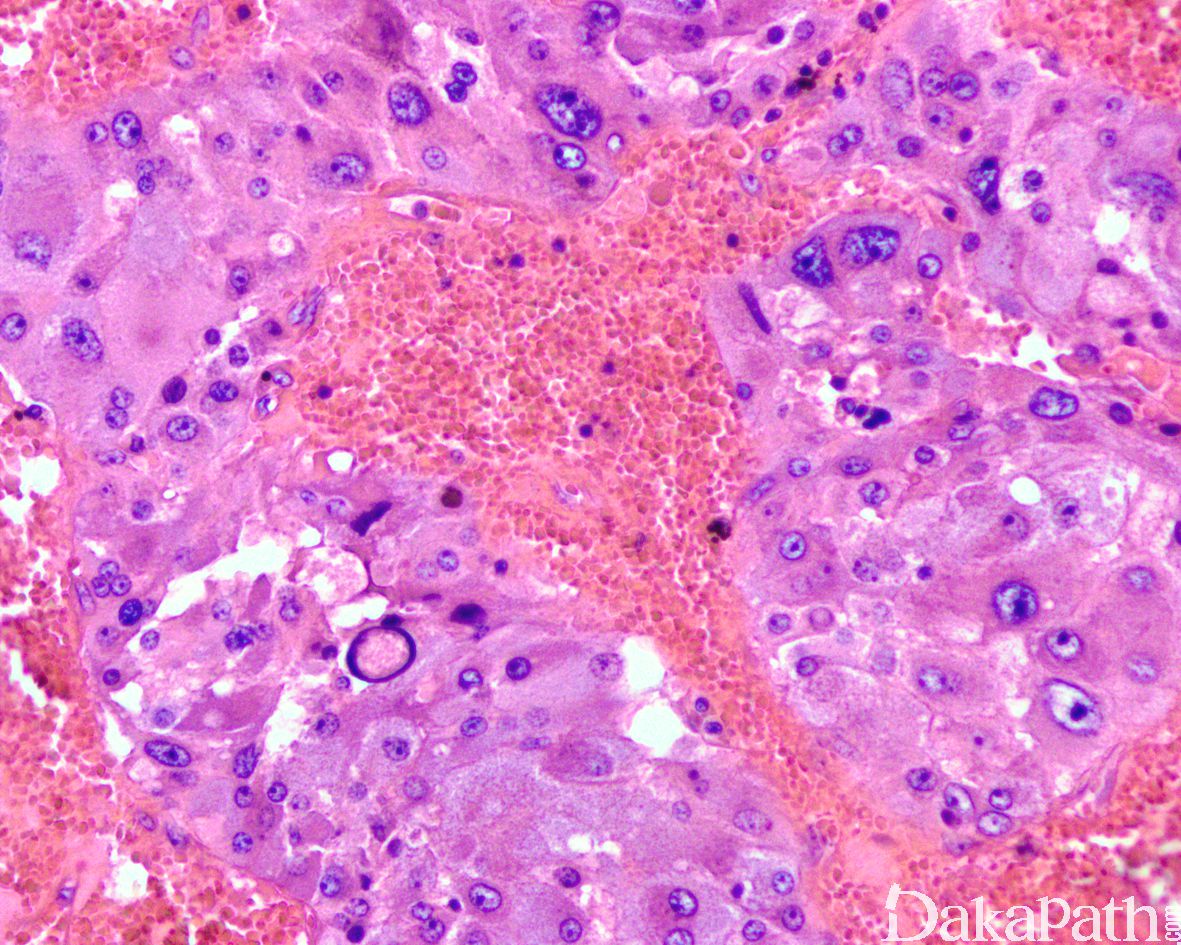
- 肿瘤边界清楚但无包膜。在横截面上,切面从浅粉红色到深红棕色不等,有时囊性变以及相关的纤维化和/或出血; 肿瘤大小不一,平均 3-6cm,部分可超过 10cm;
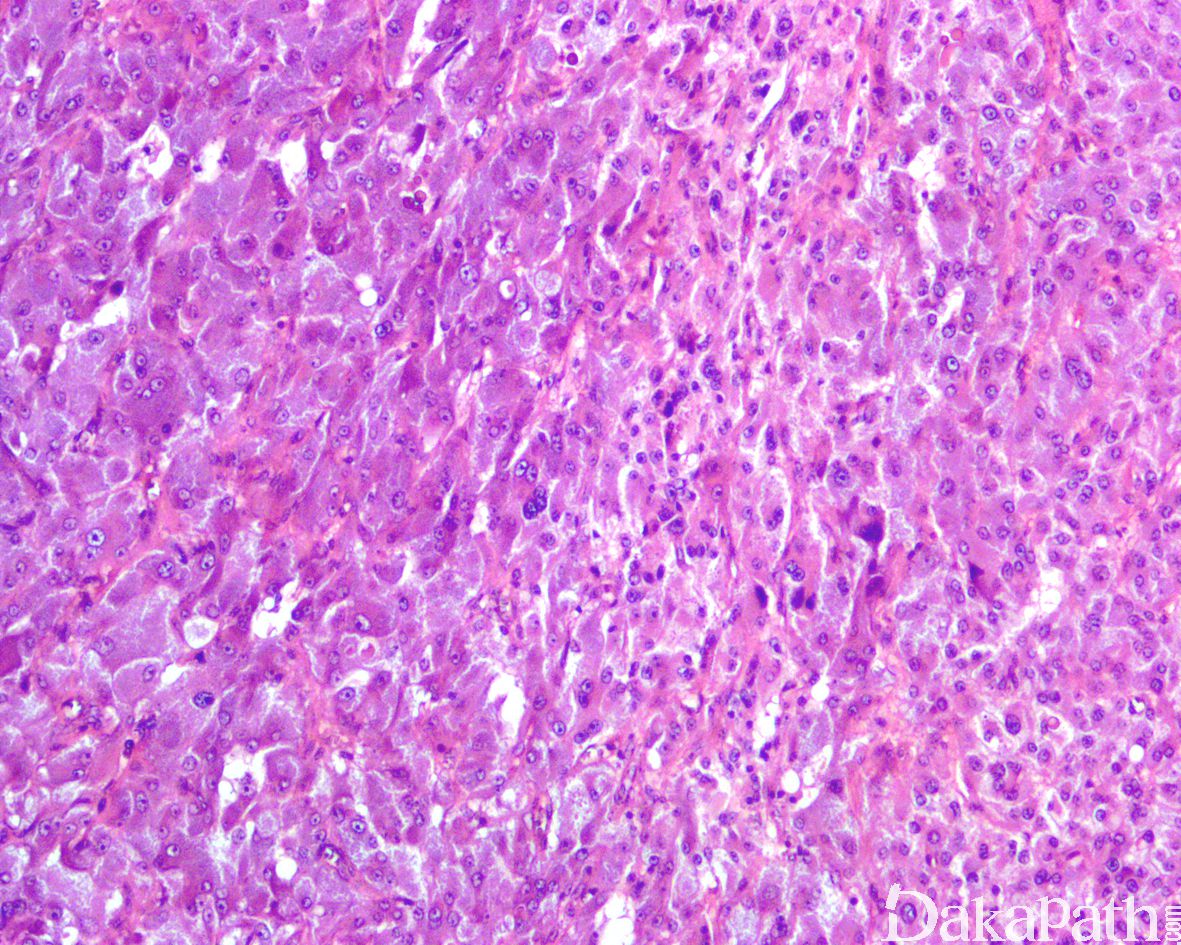
5,镜下肿瘤显示轻度的组织学异质性。大多数都具有经典的 zellballen 组织学构型,由均匀的多边形嗜铬细胞巢组成,周围环绕着支持细胞和丰富的血窦;有时可表现为明显挤压的梁状结构和弥漫性排列;
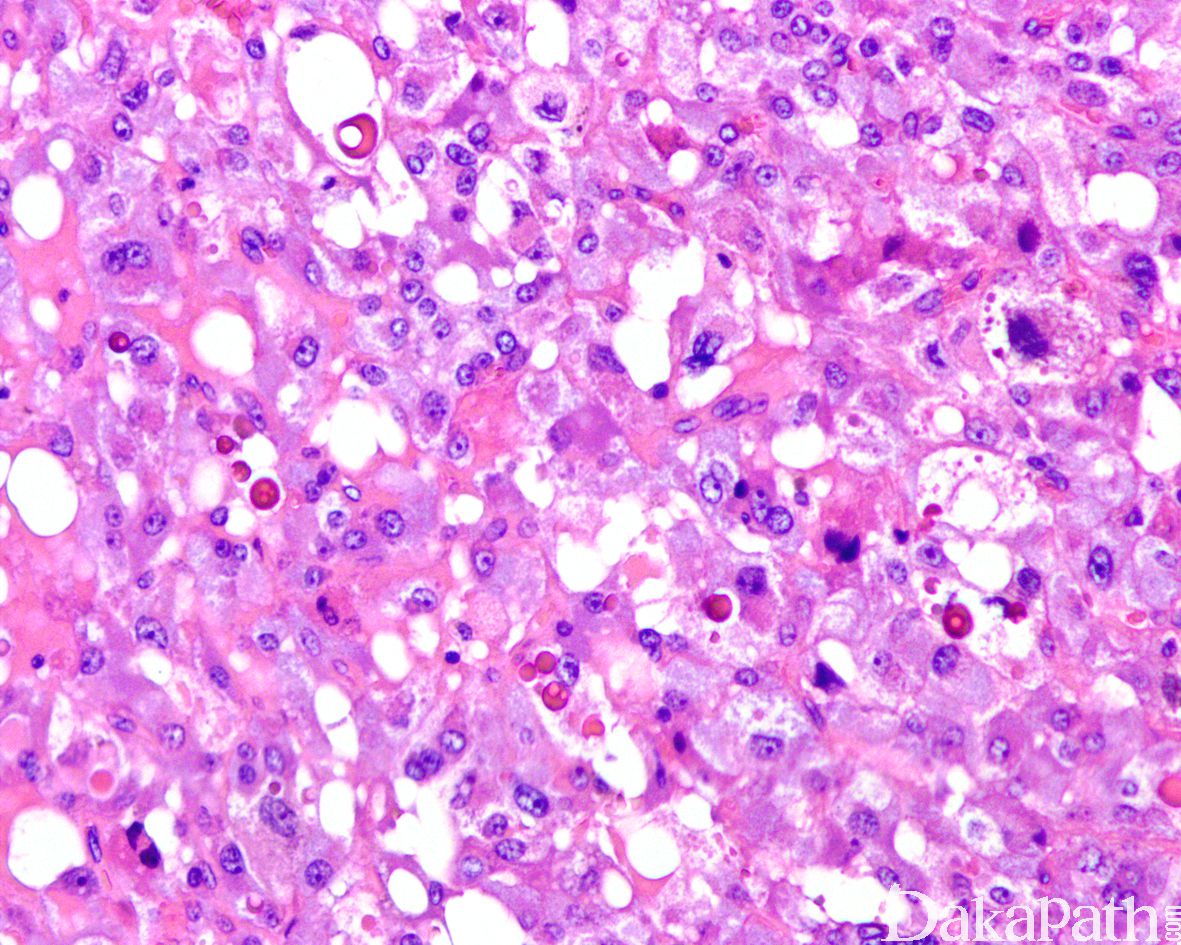
6,细胞学上,瘤细胞表现为颗粒状嗜碱性到双嗜性胞浆和空泡状核的多角形细胞,有时可见显著的核多形性和明显的核仁以及核内包涵体,偶尔可表现为一致的小圆细胞或局灶的梭形细胞,常见胞浆内外的玻璃样变嗜酸性小球,核分裂象罕见,尤其是非典型核分裂象很罕见;
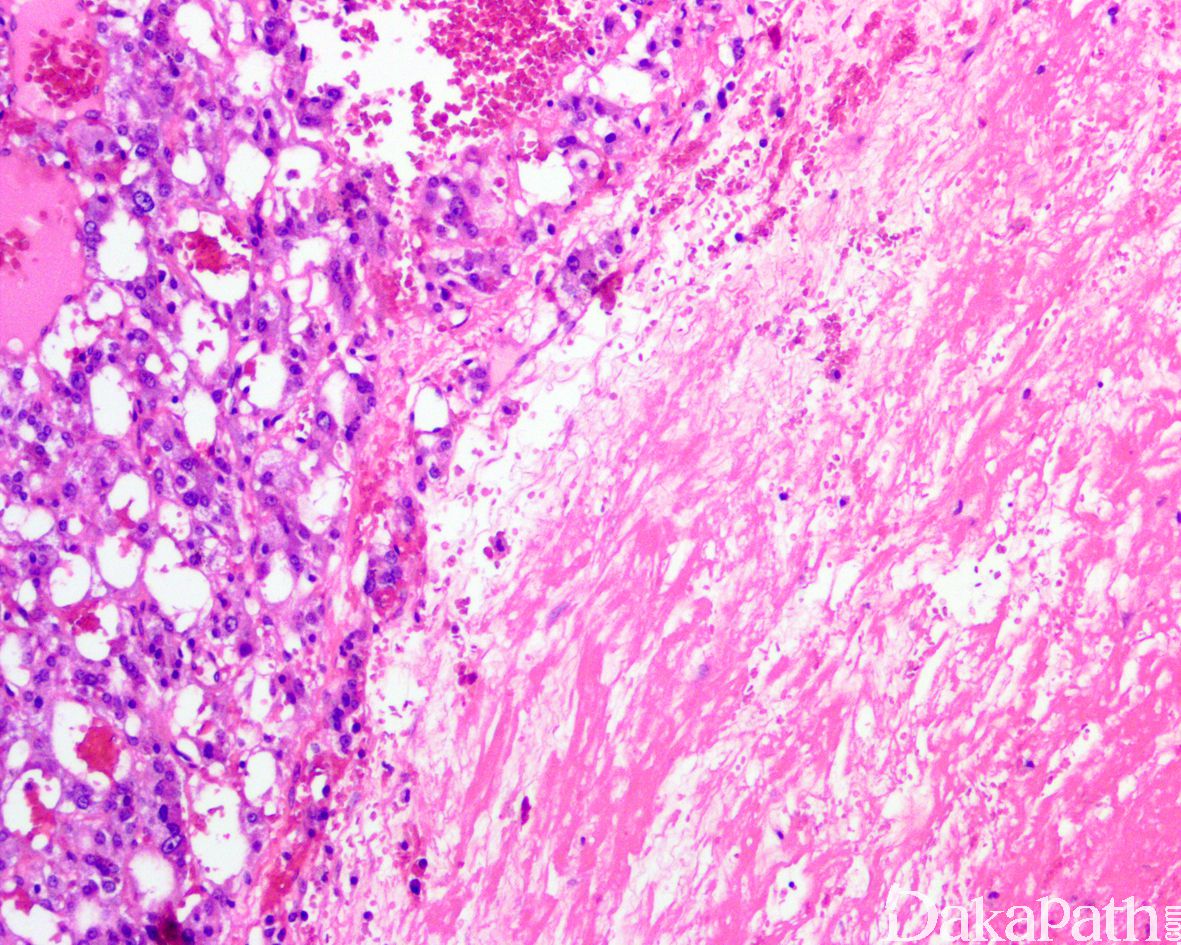
7,部分病例可表现为广泛的间质纤维化透明变和水肿,瘤细胞呈挤压的条索状排列以及微囊网状和假腺样排列。


免疫组织化学染色:
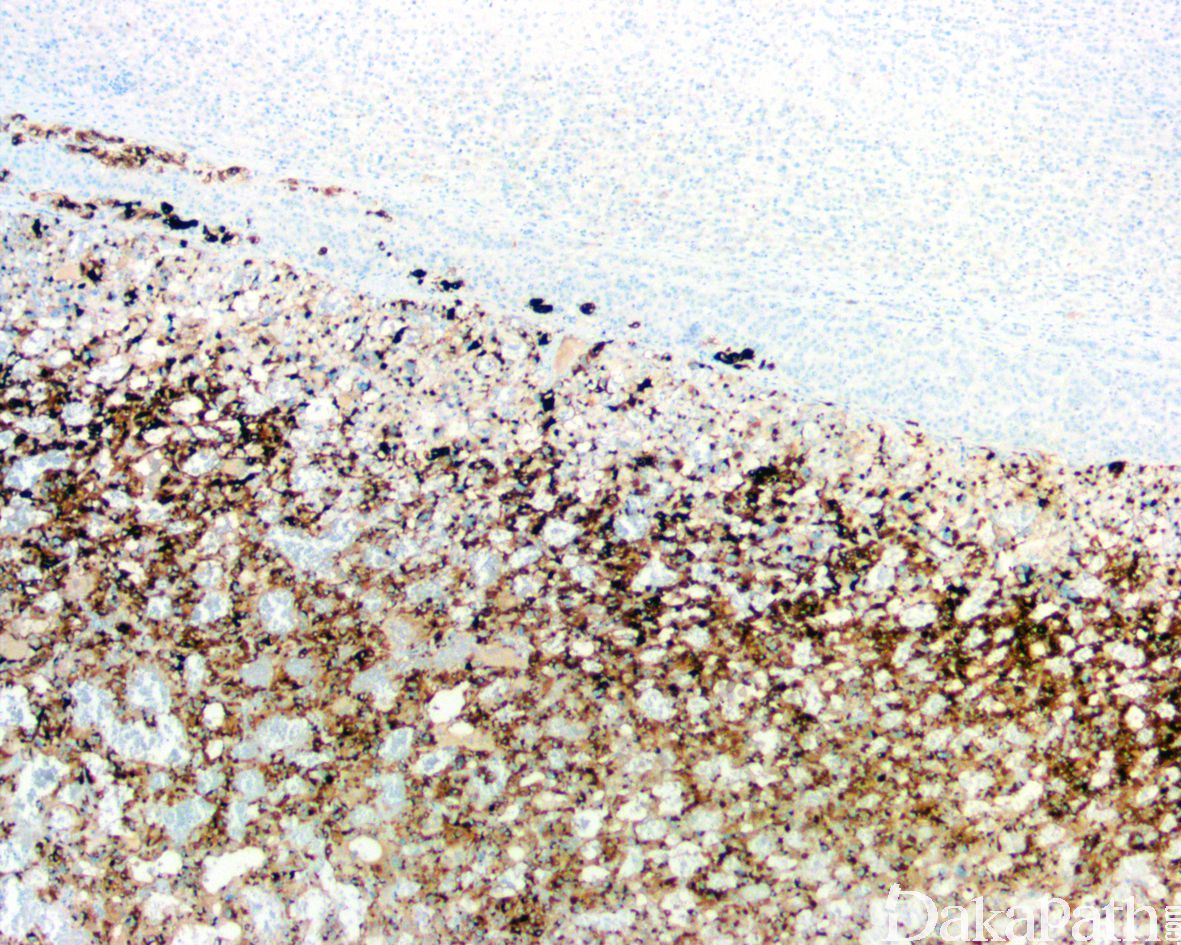
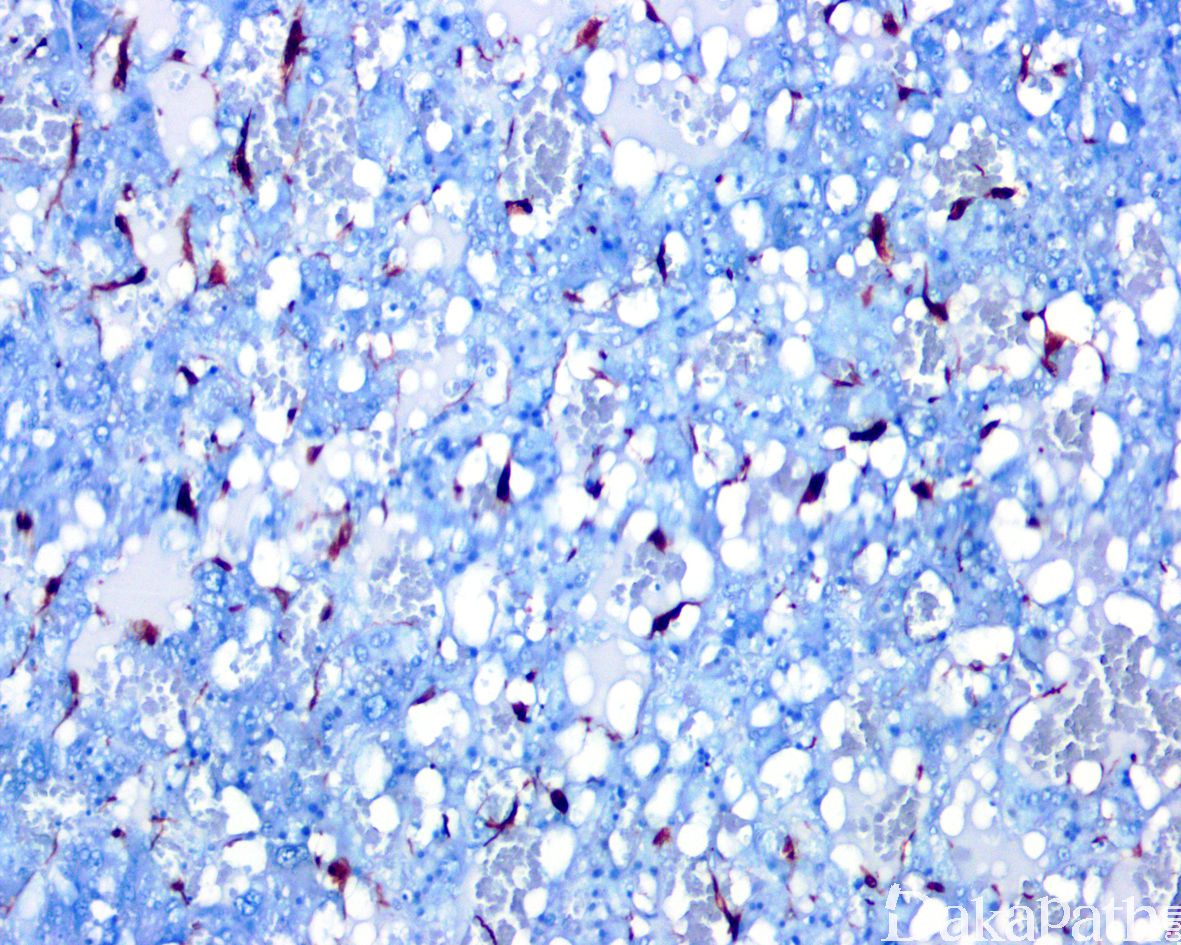
瘤细胞表达神经内分泌标志物 CD56,CGA,SYN,INSM-1 等,偶可表达 NSE 和 NFP,支持细胞表达 S100 蛋白和 GFAP,不表达肾上腺皮质标志物。伴有 SDH 缺陷者 SDHB 表达缺失。
分子标记:
嗜铬细胞瘤多数为散发性,最常见的体细胞突变为 NF1 突变,其次为 VHL 突变和 RET 突变。约 24%与遗传性综合征相关。遗传易感基因包括 NF1, ATRX Chromatin Remodeler (ATRX), HRas Proto-Oncogene, GTPase (HRAS), VonHippel-Lindau (VHL), endothelial PAS domain-contain protei
鉴别诊断:
神经内分泌癌:表达 CK,无支持细胞 S100 和 GFAP 表达
血管母细胞瘤:胞浆内富含空泡,弥漫表达 S100 蛋白、Inhibin-a,nse 等,不表达神经内分泌标志物
肾上腺皮质肿瘤:表达 calretinin,SF-1,MELAN-A, Inhibin-a 等。
预后:
所有的嗜铬细胞瘤无论组织学类型,均具有恶性潜能,均可发生转移,组织学较难预测转移的潜能。已知有几种因素会影响嗜铬细胞瘤的预后,其中儿茶酚胺分泌过多,组织学参数(如出现包膜浸润,血管浸润,非典型核分裂象和坏死等),SDHB 突变,甲基化模式,不完全切除和存在转移灶等。发生转移的嗜铬细胞瘤的 5 年总体生存率低于 60%,出现肝和肺转移至总体生存时间低于 5 年。
治疗:
完整切除,并密切随访。
参考文献:
Lloyd RV, Osamura RY, Kloppel G, et al, editors. WHO classification of the tumours of endocrine organs. 4th edition. Lyon (France): International Agency for Research on Cancer); 2017