淋巴瘤样肉芽肿病
Lymphomatoid Granulomatosis, LyG
概述:
淋巴瘤样肉芽肿病(LyG)是结外 EBV 相关的淋巴组织增殖性疾病,其特征是 EBV 阳性 B 细胞和大量混合的 T 细胞呈血管中心性和血管破坏性浸润。根据大 B 细胞数量的多少,表现出不同组织学级别和临床侵袭程度。
发病部位: 肺、皮肤、肾、肝、中枢神经系统。
诊断要点:
临床罕见,主要发生于成年人,30-40 岁,男女之比为 2:1。有免疫缺陷者(如器官移植,化疗,HIV+,先天免疫缺陷)发病危险性增加。肺是最易受累的器官,X-线见多发结节,表现为呼吸困难或胸部疼痛。可见发热、全身乏力、体重减轻等全身症状。其次最常见受累器官是皮肤,表现出皮肤结节、斑丘疹、甚至溃疡。
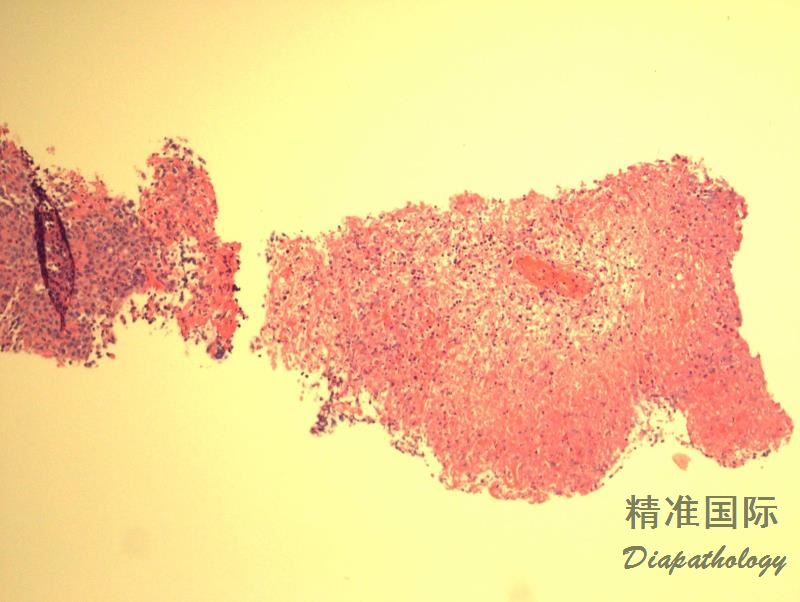
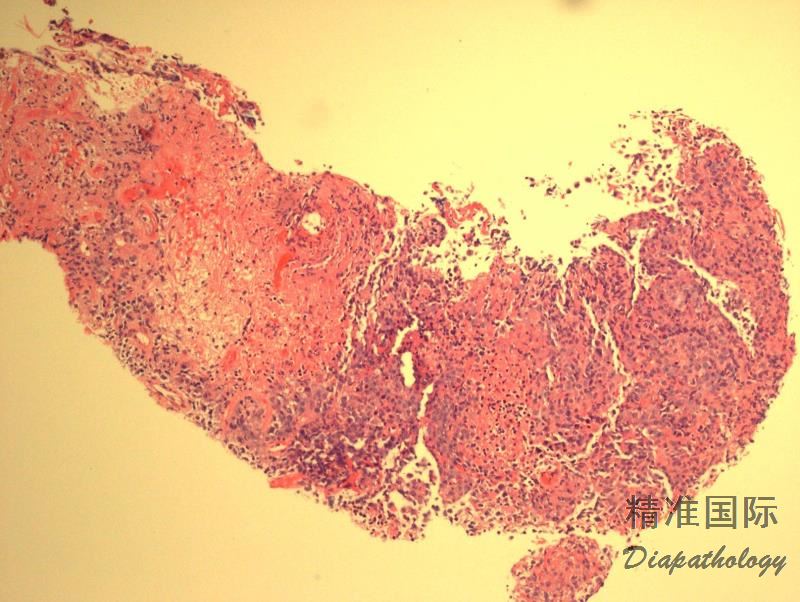
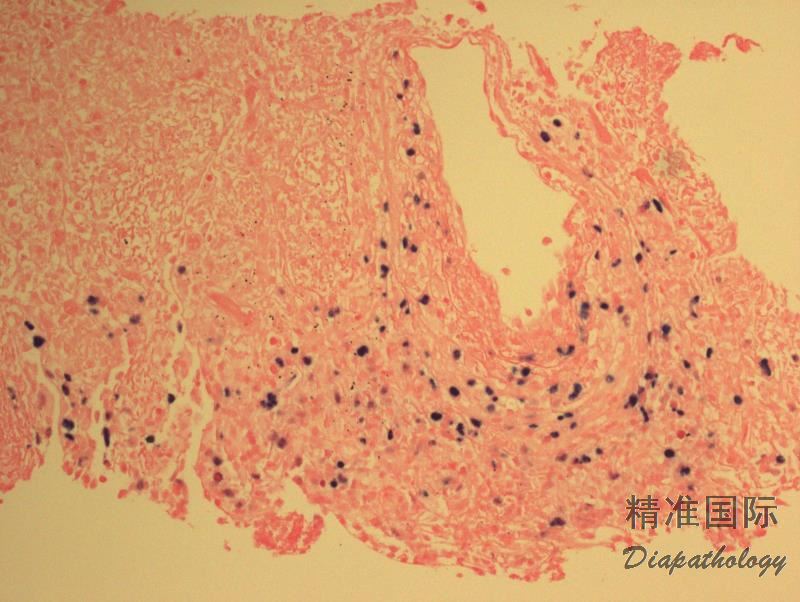
多态性淋巴细胞呈现血管中心性和血管破坏性浸润,并混有组织细胞和浆细胞,但粒细胞少见或缺如。常伴有不同程度的坏死。通常无典型的肉芽肿及其多核巨细胞(在皮肤部位则肉芽肿常见)。
浸润的淋巴细胞多数为反应性小细胞,混有不等数量的肿瘤细胞,形态不典型,核圆形或椭圆形,有核仁,可类似免疫母细胞,有时可见 HRS 样细胞。
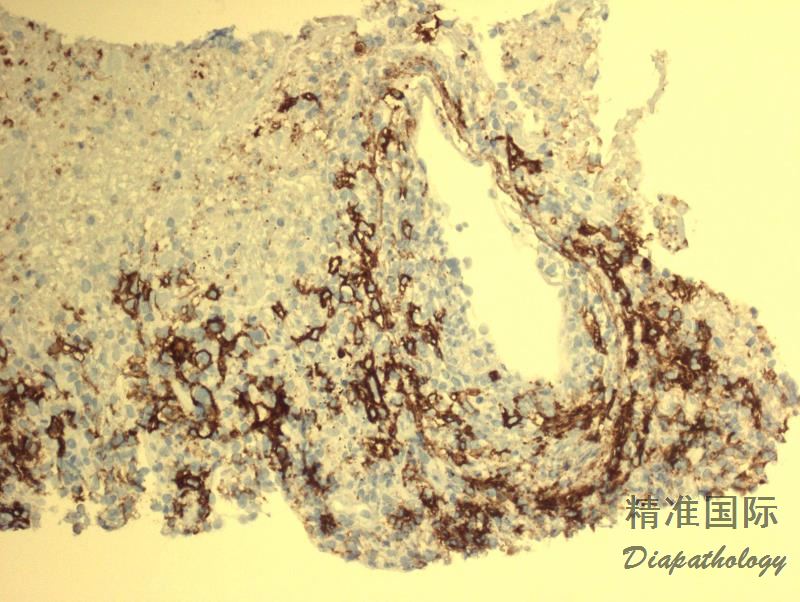
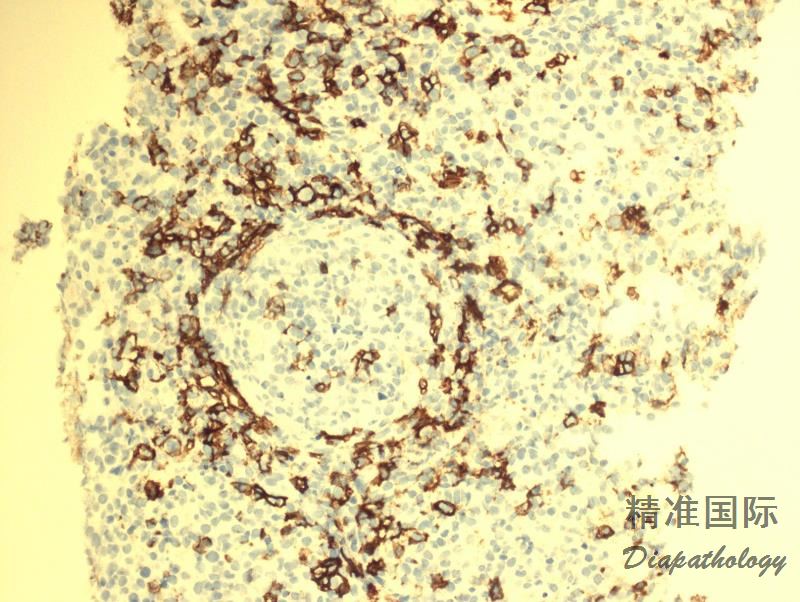
肿瘤细胞表达全 B 细胞抗 CD19. CD20. CD79a、PAX5,CD45+,CD30+/-,CD15-,EBER+;反应性小淋巴细胞表达 T 细胞抗原 CD3. CD5. CD4. CD8(CD4>CD8)。
单克隆 Ig 基因重排(2-3 级)。
分级:根据 EBV 阳性大细胞数和坏死程度分为 3 级。1 级:多态炎性细胞侵润,大细胞罕见,EBV 阳性细胞<5/高倍;2 级:多态炎性细胞背景,有时见到不典型大细胞,偶尔有小聚集(染色 CD20 易见),EBV 阳性细胞 5-20/高倍;3 级:仍有多态炎性细胞,但有较多不典型大细胞,形成大的聚集甚至局部成片,EBV 阳性细胞>50/高倍。
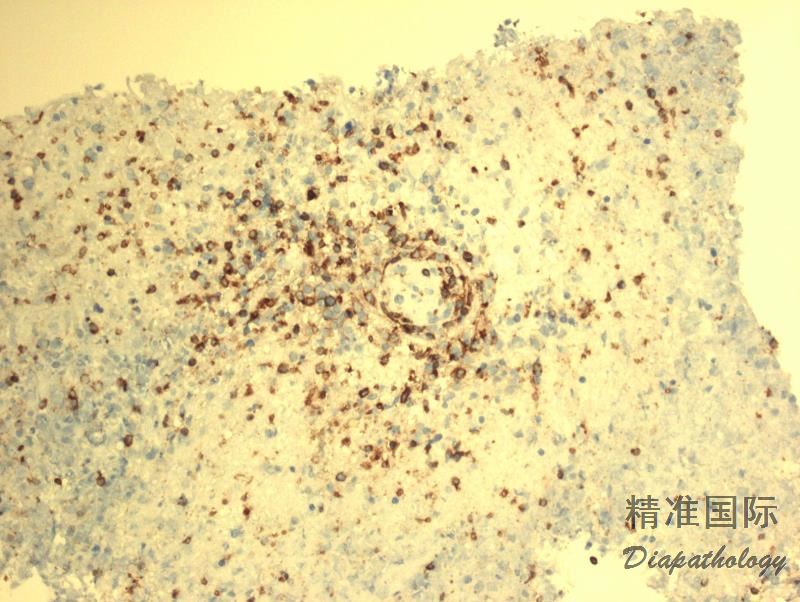
免疫组织化学染色:
肿瘤细胞表达全 B 细胞抗 CD19. CD20. CD79a、PAX5,CD45+,CD30+/-,CD15-,EBER+;反应性小淋巴细胞表达 T 细胞抗原 CD3. CD5. CD4. CD8(CD4>CD8)
鉴别诊断:
- 结外 NK/T 细胞淋巴瘤,鼻型 :弥漫非典型淋巴细胞浸润,常有坏死及血管侵犯,但多态炎症细胞少见。肿瘤细胞表达胞质 CD3. CD2. CD56. TIA1,EBER+,不表达 B 细胞抗原。
- 移植后淋巴增殖性疾病(PTLD) :与 LyG 相似,存在坏死和 EBER+ 细胞,但肿瘤表现为非典型 B 细胞弥漫侵润,细胞多形、大小不等,可伴浆细胞分化,T 细胞少。有器官移植病史。
- Wegener 肉芽肿 :可出现地图样坏死,但常有肉芽肿形成和大量中性粒细胞浸润,血管中心坏死少见,无 EBV+不典型大 B 细胞。
- 外周 T 细胞淋巴瘤一非特指 (PTCL-NOS):可累及肺部、皮肤结外组织,但多有淋巴结肿大。肿瘤表现为弥漫异型 T 细胞增生,可有抗原丢失。B 细胞量少,EBER 一般阴性。克隆性 T 细胞受体基因重排阳性。
- 经典型霍奇金淋巴瘤(CHL) :大量炎性细胞胞包括小淋巴细胞、组织细胞、浆细胞和嗜酸性粒细胞,混有散在的 HRS 细胞(有时可形成松散聚集),表达 CD30. CD15,但 CD79a 一般阴性,CD20 阴性或部分细胞不同程度的阳性,EBER-/+。可有局灶坏死,但无血管中心性坏死倾向。
预后:
变化很大,从惰性表现到侵袭性淋巴瘤。经现代治疗,总体 5 年生存率可达 70%。